
|
|||||

Tracks 1-18 |
||||||||||||||||||||||||||||||||||||||
|
Select Excerpts from the Interview
Tracks 3-4
![]() DR VOGELZANG: It turns out that the VHL gene product is an ubiquitination
enzyme. A protein called an ubiquitin protein marks cellular proteins
for destruction. You put a string of ubiquitins on a protein, and it’s basically
flushed down the toilet — the toilet being the proteosome. The VHL protein
is an ubiquitination ligase. It ties an ubiquitin protein to other proteins.
DR VOGELZANG: It turns out that the VHL gene product is an ubiquitination
enzyme. A protein called an ubiquitin protein marks cellular proteins
for destruction. You put a string of ubiquitins on a protein, and it’s basically
flushed down the toilet — the toilet being the proteosome. The VHL protein
is an ubiquitination ligase. It ties an ubiquitin protein to other proteins.
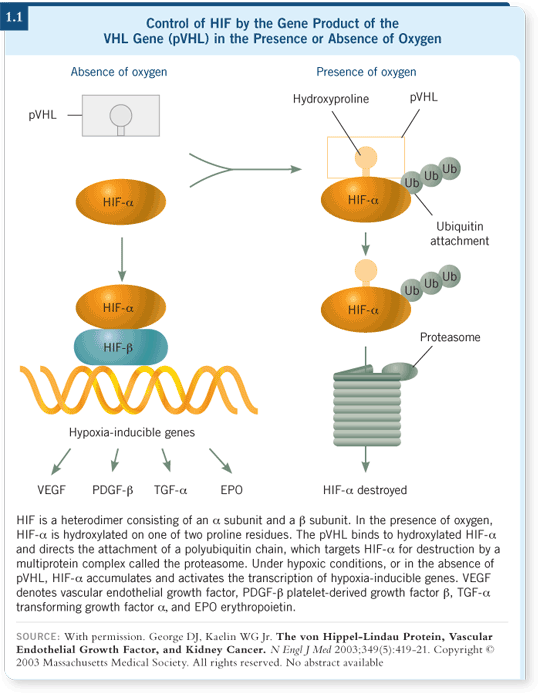
Primarily, it ubiquitinates hypoxia-inducible factor, or HIF. HIF protein levels increase under hypoxic conditions.
What does HIF do? It is an important protein and gene — it controls nearly 200 proteins, which regulate activities such as glucose transport, erythropoietin production and VEGF production (1.1).
In a hypoxic environment, erythropoietin and hemoglobin are increased and more glucose transport is produced. Likewise, under the conditions of an abnormal VHL, HIF is not eliminated. It sticks around, and instead of being flushed down the toilet, HIF protein increases; it’s not targeted for destruction. HIF becomes more active and produces these 200 or so proteins that it controls; one of them happens to be VEGF.
We now know that of the clear cell tumors — 70 to 80 percent of all kidney cancers are clear cell — a majority are VHL mutated. Therefore, it is probable that all these new agents — sunitinib, sorafenib and anti-VEGF drugs — only are working on those kidney cells that produce HIF and HIF-driven proteins. The nonclear cell tumors and perhaps some of the clear cell tumors are not producing the HIF-driven proteins.
We could group clear cell tumors into two categories: VHL mutated and VHL nonmutated. However, we don’t have an easy way to quickly measure that in the blood or in the protein. The hypothesis is that the very well-differentiated renal cells will be responsive to these agents and the very poorly differentiated, aggressive tumors will not (Teh 2006).
Track 5
![]() DR VOGELZANG: Let’s go back to the story of the von Hippel-Lindau
mutation followed by HIF overexpression and overexpression of some 200
proteins. Why did VEGF inhibitors work? VEGF is one of the major factors
that is upregulated by HIF, but EGF, PDGF and glucose transporters are also
upregulated, and all of these factors provide targets.
DR VOGELZANG: Let’s go back to the story of the von Hippel-Lindau
mutation followed by HIF overexpression and overexpression of some 200
proteins. Why did VEGF inhibitors work? VEGF is one of the major factors
that is upregulated by HIF, but EGF, PDGF and glucose transporters are also
upregulated, and all of these factors provide targets.
Jim Yang was on the team with Bill Kaelin, who developed the HIF story (Hoffman 2001; George 2003), and he evaluated a VEGF inhibitor in a placebo-controlled trial (Yang 2003). Results from his study indicated that low-dose and high-dose bevacizumab definitely slowed the cancer down. Not many PRs were present, but the cancer slowed down.
This was a critical finding because it linked the molecular biology of renal cell cancer to clinical treatment. We have known for decades that renal cell carcinoma is hypervascular.
Track 6
![]() DR VOGELZANG: Clear cell carcinoma arises from the proximal tubule, which
is bathed in “awful” acidic urine. I conceptualize it as a leathery, thick skin
cell because it has to survive in an acidic, toxic environment. It’s a tough cell.
DR VOGELZANG: Clear cell carcinoma arises from the proximal tubule, which
is bathed in “awful” acidic urine. I conceptualize it as a leathery, thick skin
cell because it has to survive in an acidic, toxic environment. It’s a tough cell.
These cells also overexpress P-glycoprotein, which is the transport protein that helps get proteins from the urine back into the tissues. The kidney and kidney cancer cells vastly overexpress P-glycoprotein, which is probably why drugs like paclitaxel, doxorubicin and the epothilones don’t work and, it is my belief, why the antimetabolites work. Gemcitabine and capecitabine work, but not very well.
Renal cell carcinoma does not respond to chemotherapy, and therefore everybody pursued other agents, such as the cytokines, nontraditional chemotherapies and lately anti-VEGF agents.
Track 10
![]() DR VOGELZANG: Better-risk patients — like those who have had their kidney
removed; whose hemoglobin, calcium and LDH are normal; people who only
have one site of disease or are feeling good — have a very good survival rate,
and you could treat them with a relatively nontoxic agent. I believe sunitinib
and sorafenib are equally good, although they have not been compared yet and
they never are likely to be compared in the metastatic setting.
DR VOGELZANG: Better-risk patients — like those who have had their kidney
removed; whose hemoglobin, calcium and LDH are normal; people who only
have one site of disease or are feeling good — have a very good survival rate,
and you could treat them with a relatively nontoxic agent. I believe sunitinib
and sorafenib are equally good, although they have not been compared yet and
they never are likely to be compared in the metastatic setting.
Sorafenib has been shown to extend life in the second line; sunitinib has now beaten interferon in progression-free survival as first-line therapy but has not yet been reported as an overall survival advantage, although we all believe it will be.
I would argue that it doesn’t matter which agent you administer first.
| Terms of Use and General Disclaimer| Privacy Policy Copyright © 2008 Research To Practice. All Rights Reserved. |

